钾代谢与血钾管理
钾代谢异常与血钾管理
70kg成人总体含钾约50mmol/kg,女性由于脂肪较多,体钾总量相对减低,平均2300mmol。总钾量大部分在细胞内(肌肉细胞),小部分在细胞外液。
细胞内钾 3300mmol,98% 肌肉2500mmol 红细胞250mmol 肝脏250mmol 骨骼300mmol 细胞外钾65mmol,2% |
每天摄入的钾变化大,多从外界摄入,成人每日摄入钾2-4g,大多数人的摄入量没有达到推荐的摄入量,远低于史前人类。
正常血钾维持在为3.5-5.5mmol/L。当血钾浓度低于3.5mmol/L时,为低血钾,血浆钾浓度高于5.5mmol/L以上时为高血钾。
血钾受Na+-K+-ATP酶、血液pH、血浆中的胰岛素、肾上腺素及醛固酮等影响。血pH改变±0.1则血钾向反方向改变0.6~0.7mmol/L。代谢性酸血症、急性应激或交感神经兴奋时,可使血钾降低,并使胞内钾转移到胞外,尿钾排泄减少;代谢性碱血症时钾向胞内转移,尿钾排泄增多,血钾降低,胰岛素使胞外钾转移入胞内,体内有机酸积聚不引起高血钾,但矿盐酸积聚可致高血钾。
人体每天从肾排钾,尿钾排泄是调节血钾的重要因素。
肾滤过的钾大部分在近曲小管及髓襻重吸收,尿钾主要由远曲小管及集合管亮细胞所分泌,受醛固酮、尿pH、尿流率及膜极化所调节。上皮细胞内钾浓度高,管腔内低,浓度差远大于电位差,促钾分泌。醛固酮使Na+-K+-ATP酶活性增加,促远曲小管泌钾。远曲小管尿流量和钠浓度增加,也促钾分泌增加,襻利尿剂促进排钾。
肾对保存钾的能力远不如保钠,肾功能正常,摄入钾多,尿钾排泄增多,健康人即使数日不摄入钾,每日肾排钾仍在每天排出20-30mmol/L,肾失钾时每日尿钾排出增多,通常在30-50mol/d以上,甚至100mmol/d。当严重低血钾持续数日,尿钾甚至低于<20mmol。
摄入70- 100mmol/d | 排出 |
90%食物及饮料
| 尿80%~90%(90-95mmol/d) 肠道10%(5-10mmol/d) 出汗 |
问题 影响血钾的因素有哪些?
1. 盐皮质激素 Ald有潴钠排钾作用,导致血钾降低 2. 胰岛素:促细胞外钾进入细胞内液 3. p H :碱中毒可导致低血钾 4. 代谢因素:分解代谢导致血钾升高 5. 渗透压(渗透浓度):高渗透压,导致水从细胞外液转移,引起高血钾 |
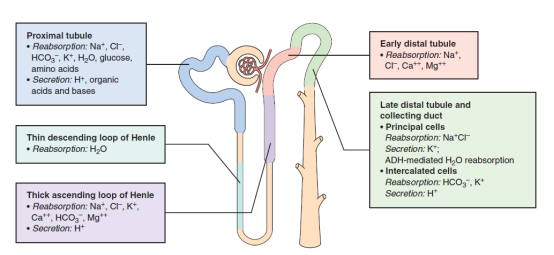
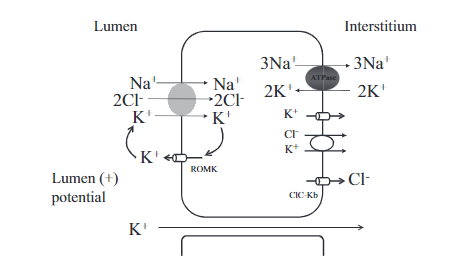
肾滤过钾绝大部分被近曲小管及髓襻重吸收。K+经肾小球全部自由滤过,在近端肾小管和髓袢升支重吸收,其吸收量占滤过量的75%,且不受钾摄入的影响。
在近端肾小管没有重吸收的钾在髓袢升支粗段通过跨细胞及细胞旁再吸收。细胞内的钾通过钠泵进入血液,也可通过K+-CL-共同转运体或CIC-Kb(氯离子通道)经基底膜侧进入血液。
近端肾小管K+重吸收 跨细胞途径(随水钠重吸收驱动),大部分 管腔正电位驱动 基底侧肾间质的k+在钠泵作用下进入细胞内,与经氯离子通道偶联(氯离子及钾离子回到血液),近端肾小管顶膜细胞的钾通道稳定正电位。 Na+-偶联葡萄糖和氨基酸的重吸收有依赖正电位 |
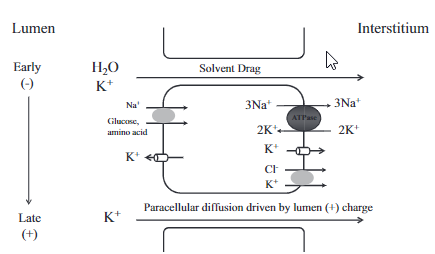
髓袢升支粗段K+重吸收 跨细胞,继发于主动吸收 Na+-K+-Cl-共同转运体,其K+由顶膜的ROMK(肾髓质外钾通道)钾通道提供 细胞旁, 腔正电位 由再循环的k+提供维持 |
问题 为什么要提高钾摄入量?
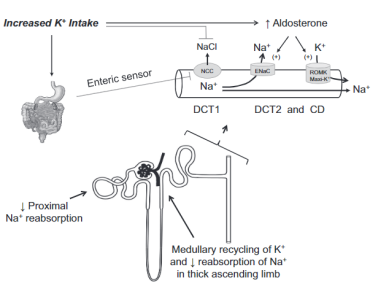
普遍低摄入,鉴于钾的重要性:降低血压、减少中风的风险,促进骨健康及减少肾结石的风险。需要提高钾摄入量。肾脏在处理高摄入钾中起重要作用,肾脏主要依靠皮质集合管(CCD)维持尿钾排泄,其次是醛固酮敏感的远端分泌肾单位(ASDN),包括远曲小管的后段(DCT2)、连接小管(CNT)。
问题 肾小管分泌钾?食物钾摄入过多肾脏如何维持低的血钾?
尿钾主要由远曲小管及集合管亮细胞所分泌(暗细胞重吸收),受醛固酮、尿pH、尿流率和膜极化所调节。
在远曲小管及集合管上皮细胞内钾很高,可达150mmol/L,而管腔内仅<15mmol/L,故其浓度差远大于电位差,促使钾分泌,醛固酮增多则Na-K-ATP酶活性增强,使远端肾小管泌钾增多,远端肾小管内尿流量和钠浓度增高,也可促使钾分泌,所以使用襻利尿剂或摄入高钠,均可增加钾的排泄。
ROMK肾外髓质钾通道是远端肾小球分泌钾的主要机制。K+再循环产生的正电位促进肾小管阳离子的吸收,钙离子及镁离子是经细胞旁离子流进入管周液,速尿作用于Na+-K+-Cl-共转运体,导致K+吸收减少,也影响钙离子及镁离子的吸收,这就是利尿剂治疗高血钙的机制。 Maxi-K+或BK通道调节K+的分泌(流量增加)。
当摄入高钾食物时,远端肾单位的转运Na+及K+的特征能够适应任何增加的细胞外液钾浓度。此时,肾小球滤过率增加和肾小管流量增加,高钠和高流量强化ROMK分泌钾的功能,钠离子转运增加(通过ENaC)激活Maxi-K+。高流量稀释钾离子浓度,有利钾分泌。所有这些都有利于避免出现高血钾。
高摄入的钾在肾间质积累会抑制近端肾小管和髓袢升支粗段的盐转运,导致远端肾小管转运的水钠增多,有利于远端肾小管k+分泌增加。
当饮食钾升高时,NCC的活性受到抑制,导致转运的钠及流量增加,到达ASDN及集合管的钠及流量增加,促进醛固酮敏感的部位分泌钾离子增加。同时由于血钾浓度变化促使醛固酮分泌增加,有利于钾分泌。ENaC的作用生电Na重吸收,K+分泌。
远曲小管 k+感受器 | DCT1()
| DCT2 |
利尿剂作用部位 | 噻嗪类敏感的NaCL共同转运体(NCC) | 醛固酮敏感(ASDN) |
问题 血钾增高如何抑制NCC?
WNK。由于血钾增加,使DCT1细胞去极化,导致细胞内的氯离子增加,激活WNK,WNK抑制NCC的活性。当血钾减低时,刚好出现相反效应,NCC激活,减少到醛固酮敏感部位的钠转运量和流量。有证据表明Kir4.1/5.1通道在此过程发挥作用。血钾增高导致Kir4.1/5.1通道兴奋,介到NCC活性改变。
当pH增加时,也有利钾分泌。肠道的改变也有利于维持钾的稳定。
Gordon综合征
该病是由Gordon于1986年首先报道的常染色体显性遗传病,又称假性醛固酮减少症Ⅱ型,临床主要表现高血压、高血钾、代谢性酸中毒而肾小球滤过功能正常。由WNK家族(MNK1—4)中WNK1和WNK4突变所致。WNK家族蛋白位于远端肾单位集合管,调控钾-氢交换和钠-氯吸收,生理状态下,WNK1抑制WNK4,而WNK4抑制位于远曲小管肾小管上皮细WNK1、WNK4突变均可增强NCC活性,导致钠重吸收增加。2012年发现两种新的突变基因KLHL3和CLU3,KLHL3突变呈常染色体显性遗传,KLHL3可呈常染色体显性或隐性遗传。研究发现KLHL3可与WNK1、WNK4结合发生泛素化从而使之降解,并可增强肾远端小管上皮细胞膜上钾通道(ROMK)的转运功能,这也可解释Gordon综合征中KLHL3、CLU3突变者出现明显高钾血症的原因。Cormick等发现导致Gordon综合征的CUL3突变后可使KLHL3降解,从而增加WNK1、WNK4表达。这两种机制共同的结果是远端。肾单位WNK1、WNK4表达增加,NCC活性增强和ROMK活性降低,钠重吸增加,钾分泌减少,导致水钠潴留和高钾血症。不同突变基因与Gordon综合征的临床表现有显著相关性,根据发病年龄和临床表现轻重可排序为:CLU3>KLHL3>WNK4>WNK1。临床表型:高血压,高血钾,高血氯代谢性酸中毒,低血浆肾素活性,低或正常醛固酮浓度。肾小球滤过率正常。
CLU3突变者发病早,绝大多数在未成年前出现高血压,表现严重高血钾和代谢性酸中毒。而WNK1患者可能在成年后期才出现高血压,临床症状相对不明显。发病机制:WNK1功能获得性突变与WNK4、KLHL3、CUL3功能丧失突变的纯效应:过度激活钠-氯共转运体(NCC)与上皮钠通道,抑制肾外髓质钾(ROMK)通道,增加钠的重吸收,减少钾的排泄。
治疗:根据分子发病机制,提出噻嗪利尿剂的靶向治疗,抑制NCC,逆转高血钾,使血压恢复正常。Gordon综合征的治疗上应限盐,低剂量NCC阻滞剂氢氯噻嗪(12.5~25mg/d)有助于血压控制,但WNK1基因突变者对噻嗪类利尿剂敏感性相对较差。
高钾血症
高钾血症是危及慢性肾脏疾病与终末期肾脏疾病患者生命的最常见的电解质紊乱类型。随着慢性肾脏疾病患者的肾功能水平逐渐恶化,其发生几率也逐渐增加。
(1)肾排钾减少: 急性肾功能衰、慢性肾功能衰竭 皮质激素功能减退(醛固酮减少症) (2)细胞内钾移出细胞外 组织损伤 糖尿病DKA 缺氧 (3)钾摄入过多
| 高钾血症 轻度(5.1-6.0mmol/L) 中度(6.0-7.0mmol/L) 重度(≥7.0mmol/L)。
|
引起血钾增高的因素
除肾功能恶化外,以下的因素都是引起或加重高钾血症的原因:含钾饮食摄入过多、胰岛素缺乏、代谢性酸中毒、组织破坏(溶血、横纹肌溶解、肿瘤溶解以及组织缺血)、药物因素所致肾脏排钾减少:包括使用血管紧张素转化酶抑制剂(ACEI)、血管紧张素受体阻滞剂(ARBs)、盐皮质激素受体拮抗剂、保钾利尿剂、钙调磷酸酶抑制剂等。其他少见的低肾素低醛固酮血症,假性醛固酮减少症(PAHI,PAHII,Gordon综合征),IV型肾小管酸中毒(醛固酮减少或抵抗)。
当肾功能受损时,尿钾排出减少,如果摄入钾超过肾排出能力可导致高血钾。含钾食物来源广泛存在于动植物食物中。
含钾高的食物 各种干货: 紫菜、菇类、干枣、百合 多数蔬菜:菜花、油菜 各种肉类 薯类及粗粮类
| 含钾低的的食物(<250mg/100g) 大米、面粉、小米、玉米、豆腐、胡萝卜、青菜、白萝卜、大白菜、油菜心、圆白菜、冬瓜、空心菜、黄瓜、苦瓜、西红柿、绿豆芽、蒜苗、柿、柚、柑、桃、荔枝、牛奶、洋葱、南瓜、菜瓜、西瓜、葡萄、鸭梨、苹果、鸡蛋、鸭蛋、海参等食物。 |
高血钾有严重的后果,慢性肾脏病患者必须注意血钾,检测血钾。Ckd四期以上,易导致高钾血症,CKD患者避免使用高钾食物,对于接受透析的患者,摄入量为50-70mmol/d。非透析,每天钾1200-1600mg/d(1-2g)。
在低钾饮食中应少摄入富含蛋白质的瘦肉、鱼、虾、豆类食品,和浓的汤、果汁;尽量选用每百克食物中含钾低于250mg的菜蔬和水果;高钾食物可以通过浸泡或水煮的方法去除,这样可以与低钾的蔬菜及水果搭配使用,以保证必需的营养物质及生物活性物质。将食物浸泡水中,或加水煮,熬菜去汤,都可以减少其中钾的含量,这样可以与低钾的蔬菜及水果搭配使用,以保证必需的营养物质及生物活性物质。
低钠盐含钾量高,不适合CKD使用。为了管理血钾,我们需要个体化的食物清单。
食物 | 热量 | 蛋白质 | 钾 | 磷 | 钠 | 钙 | 嘌呤 |
纯牛奶 | 54.0 | 3.0 | 115.0 | 73.0 | 37.2 | 104.0 | - |
羊奶 | 59.0 | 1.5 | 135.0 | 98.0 | 20.6 | 82.0 | - |
酸奶 | 72 | 2.5 | 150.0 | 85.0 | 39.8 | 118.0 | -
|
核桃仁 | 627 | 14.9 | 161 | 894 | 6.4 | 56 | - |
甘薯 | 99 | 1.1 | 177 | 39 | 28.5 | 23 | 2.6 |
土豆 | 76 | 2 | 179 | 40 | 2.7 | 8 | 2.6 |
大米 | 346 | 7.4 | 122 | 110 | 4.3 | 41 | 18.4 |
淀粉 | 345 | 1.2 | 142 | 25 | 6.3 | 18 | - |
玉米 | 335 | 8.7 | 130 | 218 | 3.3 | 144 | 9.4 |
黄瓜 | 15 | 0.8 | 256 | 24 | 4.9 | 24 | 14.6 |
番茄 | 19 | 0.9 | 280 | 2 | 5 | 10 | 4.6 |
苹果 | 45 | 0.7 | 378 | 11 | 0.7 | 3 | 1.3 |
梨 | 43 | 0.2 | 355 | 14 | 1.5 | 4 | 1.1 |
葡萄 | 43 | 0.5 | 989 | 13 | 1.3 | 5 | 0.9 |
冬瓜 | 11 | 0.4 | 305 | 12 | 1.8 | 19 | 2.8 |
生菜 | 13 | 1.3 | 306 | 27 | 32.8 | 34 | - |
花菜 | 24 | 2.1 | 312 | 47 | 31.6 | 23 | 25 |
鸡蛋 | 144 | 13.3 | 154.0 | 130.0 | 131.5 | 56.0 | 6.3 |
猪肉 | 143 | 20.3 | 204 | 189 | 59.4 | 6.0 | 132.6 |
茄子 | 21 | 1.2 | 272 | 2 | 5.4 | 24 | 14.3 |
大白菜 | 15 | 1.4 | 185 | 58 | 44.2 | 39 | 12.6 |
青椒 | 22 | 1 | 281 | 2 | 3.3 | 14 | 8.7 |
香蕉 | 91 | 1.4 | 351 | 28 | 0.8 | 70 | 1.2 |
胡萝卜 | 37 | 1 | 178 | 27 | 71.4 | 32 | 8.9 |
柠檬 | 35 | 1.1 | 787 | 22 | 1.1 | 101 | 3.4 |
白萝卜 | 20 | 0.9 | 178 | 26 | 618.8 | 36 | 8.9 |
高钾血症的治疗
紧急情况下使用葡萄糖酸钙拮抗,严重高钾血症需要血液透析治疗。高钾血症往往会限制一些治疗药物的使用,如肾素-血管紧张素-醛固酮(RAAS)阻断剂。由于高血钾的后果,患者被迫暂停用药。
高钾血症的治疗: 限制钾的摄入 停用引起高钾的药物如RAAS阻断剂 应用襻利尿剂或噻嗪类利尿剂 口服降钾树脂(聚磺苯乙烯钠/钙)。
|
降钾树脂其疗效被证明具有一定的降钾效果,但其应用时具有发生致死性结肠坏死及消化道穿孔等副作用的风险。
Veltassa@由patiromer阴离子和山梨醇钙反离子构成,为新型降钾药物,用于非透析和非急性的CKD患者的高钾血症治疗。由2-氟-2丙烯醇、二乙烯苯和1,7-辛二烯构成的聚合物,它是一种钙-钾交换聚合物,该药不被机体吸收,其在胃肠道内失去钙离子,与K+结合,并通过粪便排出。通过增加粪便中钾的排泄,降低消化道中游离钾的浓度,从而降低血钾水平。在健康个体,Veltassa可以降低尿钾、钠、磷、镁,并增加尿钙,且具有剂量依赖性。其主要的不良反应包括:轻到中度的便秘及轻度的低镁血症,同时可以降低患者的血醛固酮水平和血压。因此,Veltassa可能还可以通过降低血醛固酮水平,具有长期的器官保护作用。与其他药物间隔服用2-4小时。
环硅酸锆钠是非吸收性的硅酸锆,微孔大小和组成模拟生理性钾离子通道,对钾离子具有高度亲和力,可以在胃肠道捕获钾离子的能力,以氢及钠置换钾离子,与钾离子结合,并经大便排出,降低游离钾离子浓度,从而降低血钾水平。
Lokelma为口服混悬液,每包5g或10g,推荐剂量为10g,每天3次,持续48h,对于维持治疗,推荐剂量为每天1次,10g,根据需要每隔1周调整剂量(每天5g),以获得所需要的血清钾目标范围。
适应证Lokelma是一种钾黏合剂,适用于治疗成人高钾血症,因Lokelma的起效时间延迟,不应用于危及生命的高钾血症的紧急治疗。不良反应Lokelma最常见的不良反应为轻度至中度水肿。

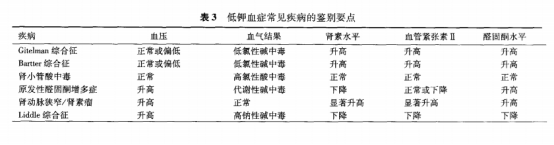
低钾血症
钾缺乏是指机体总钾量的丢失,与低钾血症并不完全一致。如临床上虽然钾缺乏,但钾从细胞内转移至细胞外或者血液浓缩,血清钾浓度可正常或增高;也可因血液稀释或钾转移到细胞内,导致血清钾降低,而体内总钾不低。
低钾血症的原因很多,最常见原因为钾的丢失,部分原因是钾的摄人量减少或者从细胞外液向细胞内转移,大面积烧伤、放腹水、腹腔引流、腹膜透析及不适当的血液透析等情况时也会出现钾缺乏。
异常丢失 药物:利尿剂、润肠通便药物、可的松 胃肠道 尿路排泄增加: 渗透性利尿、盐皮质激素过多、肾小管酸中毒(I型和II型)、多尿、肾转运钾缺陷 低镁血症 血液透析 跨细胞转运 药物:胰岛素过量使用,β2受体激动剂、Decongestants、嘌呤类似物(茶碱类)、二性霉素B 碱中毒 再喂养综合征 低体温 甲状腺功能亢进 家族性周期性麻痹 心肌缺血 头部损伤 摄入不足 厌食、禁食、认知障碍(痴呆)、肠外营养 假性低血钾 标本延迟分析
|
患者因长期禁食、昏迷、消化道梗阻或者神经性厌食以及偏食等均可致钾的摄人不足。短期内摄人不足不易引起低钾血症,通常每日氯化钾摄入量<3g,并持续2周以上,才可发生低钾血症。消化液含有丰富的钾,胃液含钾为14mmol/L,肠液含钾为6.2—7.2mmol/L,长期大量呕吐、腹泻、胃肠引流及造瘘等均可造成胃肠道失钾。
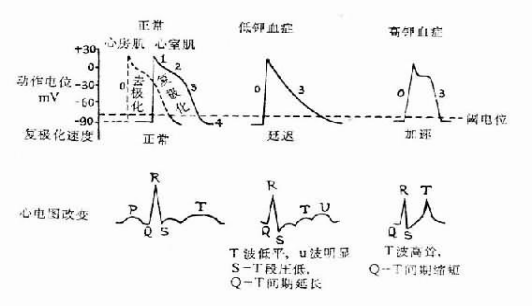
低钾血症的临床表现缺乏特异性:乏力、疲倦、肌痉挛、肌无力、麻木、恶心、呕吐、腹部痉挛、腹胀、便秘、心悸、尿量多、口渴、头晕。低血钾症的心电图表现。
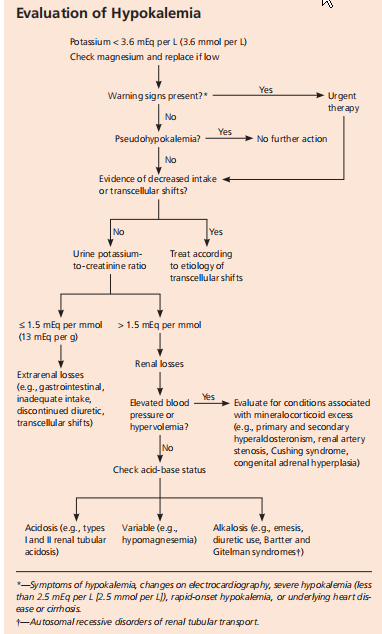
诊断流程
通过病史、体格检查并结合生化、心电图检查,确定低钾血症,从中发现病因线索。稀释性低钾血症因细胞外液潴留而稀释,血钾浓度相对降低,机体总钾量和细胞内钾可正常。见于水过多和水中毒,或过多、过快补液而未及时补钾的情况。病因分析最为复杂是钾经尿路排泄过多,其他原因引起的低钾血症易识别。进行鉴别诊断时,可以从以下3方面进行:
①测定24h尿钾的排泄以区分是否为肾性失钾;
②根据是否伴有高血压并结合血浆肾素和醛固酮水平和酸碱平衡进一步鉴别;
③结合临床和实验室检查综合判断低钾血症的病因。
问题 尿钾的测定常用的方法有哪些?
①24h尿钾排出量:尿钾在15mmol/24h以上,提示肾性失钾。
②尿钾浓度:如尿钾>20mmol/L,则多为肾脏失钾,但是<20mmol/L并不能完全排除肾脏失钾,特别对于摄钠低和药物(如呋塞米、氢氯噻嗪、乙酰唑胺等促排钾利尿剂)所致尿钾排出过多。
③尿钾/尿肌肝比值(K/C):随机尿液的钾与肌酐比值较易获得,若二者比值>1.5(mmol/mmo1),则提示肾脏失钾可能。如二者比值<1.5(mmol/mmo1)提示非肾源性失钾,如钾摄入减少、胃肠道丢失钾或钾向细胞内转移等。转移性低钾血症包括家族性周期性麻痹、甲亢伴周期
性麻痹、特发性周期性麻痹在内的多种周期性麻痹。
问题 低钾血症的心电图表现?
T波降低或倒置, u波增高,Tu融合 ST段下移 Q-T间期延长 P波增高 多种异位心律失常
|
问题 低血钾症的心电图变化机制
细胞外钾离子浓度降低时,细胞膜对钾的通透性减少,细胞内外钾离子浓度差更加明显,因此钾离子流出速度减慢,3期时间延长。由于这一作用在普肯野氏纤维较心室肌更为明显,因而动作电位的延长程度普肯野氏纤维超过心室肌,这也是低血钾时QT延长、T波低平,U波明显的原因(许多专家认为U波是普氏纤维复极的表现),此时T-U融合呈驼峰状,QT常不易测量。
血钾低时,钾离子流出减慢,4期钠离子内流增强,因此起搏细胞舒张期除极速度增加,可以使非自律细胞变成自律细胞,所以低血钾引起自律性增加,可出现各种异位心律。
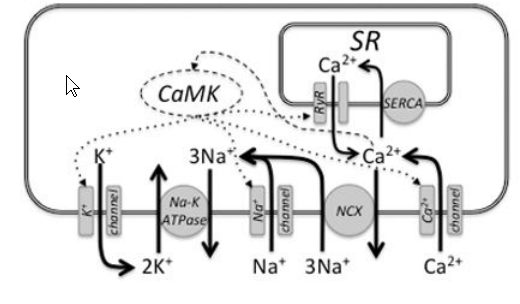
问题 低钾血症导致心律失常的机制?
1. 静息电位过度极化
2. Na+-K+ATPase抑制
3. K+通道受抑制影响钾外向电流,从而延长动作电位时程,,药物也可以抑制钾通道
导致复极化贮备减少,EAD,DADs和自律性增加。可以引起Tdp,VT,甚至室颤
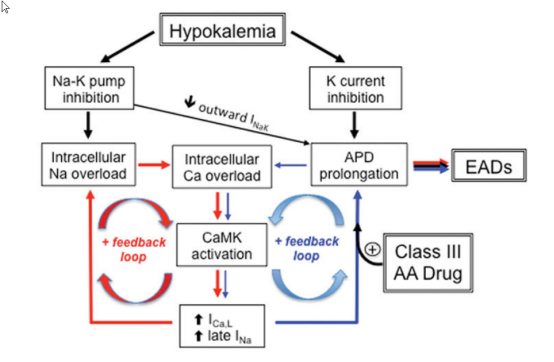
从尿路丢失钾的疾病?
包括肾脏疾病和盐皮质激素增多或具有盐皮质激素作用的激素增多。
从尿路丢失钾 |
由于肾小管病变,肾排钾过多 肾消耗病 (NPHP) 肾脏疾病急性肾衰竭多尿期 尿路梗阻解除后利尿 肾小管性酸中毒I、II及III 家族性原发性失钾性肾炎 Fanconi综合征 Liddle综合征 Bartter 综合征 Gitelman 综合征 低镁血症 盐皮质激素及盐皮质激素样作用的激素过多 药物、其他 渗透性利尿:甘露醇、高血糖
|
内源性盐皮质激素过多见于柯兴氏病、原发性醛固酮增多症、肾上腺癌、异位促肾上腺皮质激素综合征、先天性肾上腺增生(11-β羟化酶和17-α羟化酶缺乏)、GRA(糖皮质激素可治疗高血压)、肾动脉狭窄、恶性高血压、血管炎(肾供血不足)、11β-羟化类固醇脱氢酶缺乏。外源性盐皮质激素过多:激素治疗,甘草和中草药制剂含有甘草酸抑制11-β类固醇氢酶。
问题 多种药物可导致低钾血症,有哪些?
排钾性利尿药如呋塞米、氢氯噻嗪、乙酰唑胺等;渗透性利尿药如甘露醇、山梨醇及高渗糖液等;高血糖状态和补钠过多等均可使钾排出增多;
导致低钾血症药物
|
1. 细胞内外转移 肾上腺素、支气管扩张、茶碱、咖啡因、维拉帕米中毒、胰岛素 2. 尿路丢失 利尿剂 盐皮质激素及盐皮质激素样作用的药物 氟氢可的松 甘草制剂 青霉素类(羧苄西林)、碳酸氢钠、二性霉素B、庆大霉素、多粘菌素B顺铂 3. 肠道排钾 泻剂 |
问题 羧苄西林如何导致低钾血症?
因为药物改变了肾小管上皮细胞内的电位差,有利于钾的排出。
问题 低镁血症导致血钾血症的机制?
低镁血症时,由于Na一K一ATP酶失活,肾脏的保钾作用减弱(肾小管重吸收
K减少),尿排钾过多,导致低钾血症
肾小管性酸中毒(RTA)是一个综合征,常见于慢性活动性肝炎、肝硬化、原发性胆汁性肝硬化、干燥综合征、高球蛋白血症、红斑狼疮,肾脏病如肾盂肾炎、梗阻性肾病、小管间质性肾炎及慢性肾衰竭等。药物引起的,如非甾体抗炎药、碳酸锂及二性霉素B等。此外,应引起广泛重视的地方性中毒病,如棉酚和钒酸盐中毒。RTA 一般分为四型,
远端肾小管性酸中毒(Ⅰ型) 近端肾小管性酸中毒(Ⅱ型) 混合型肾小管性酸中毒(Ⅲ型) 高血钾型肾小管性酸中毒(Ⅳ型)
|
前三型均可发生低血钾症。
Ⅰ型dRTA约占RTA病例的70%,主要为H-泵障碍,出现低血钾高血氯性酸中毒,高尿钙、低血钙和低血钠,继发甲旁亢及高尿磷和低血磷,尿铵生成减少,尿pH>5.5。
ⅠⅠ型pRTA为H+-Na+泵障碍,H-Na不能交换,则尿HCO3排出增多(正常时滤液HCO3约85%在此处重吸收),发生低血钾高血氯性酸中毒,近小管对HCO3重吸收是由上皮顶膜的H-Na泵和H-泵把H排入管腔,经胞膜的碳酸酐酶作用与HCO3生成CO2和H2O,又扩散入胞内再经碳酸酐酶作用生成H和HCO3;后者经基侧膜的N-HCO3泵和Cl-HCO3泵再排出胞外。此外,肾小球滤过的葡萄糖、氨基酸、磷酸盐及尿酸等在近小管内重吸收是与钠重吸收相耦联,与Na-K-ATP酶有关,钠不能重吸收则这些物质也不被重吸收,出现于尿中,称为Fanconi综合征。
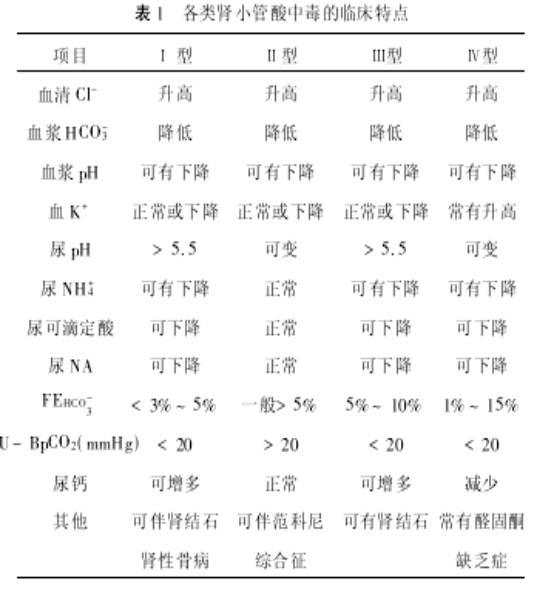
问题 Fanconi综合征
分为原发性及继发性两大类。原发性多为先天性肾小管功能缺陷,常与遗传有关,该病人生长发育正常,原无明显的疾病史,故可能性不大。继发性多由肾小管间质疾病、特别是慢性间质性肾炎引起。慢性间质性肾炎常见病因有:
①细菌、病毒、真菌感染;
②理化因素损伤:
③代谢性疾病(高尿酸血症、高钙血症)
④免疫性疾病:
转移性低钾血症
碱血症 输注大量葡萄糖液 周期性瘫痪: 其它 |
细胞内钾是细胞外钾的30–40倍。这主要由钠钾ATP泵维持。在病理情况下,细胞外钾转移至细胞内,其特点是体内总钾量正常,细胞内钾增多,血清钾浓度降低。
(一)碱血症:一般pH每升高0.1,血清钾约下降0.7mmol/L。通常呼吸性碱中毒影响较小,代谢性碱中毒时细胞外液H浓度降低,细胞内H+释放出来,而细胞外液中K+进入细胞,发生钾分布异常。此外,碱中毒时肾小管上皮细胞排H+减少,故Na+-H+交换减少而Na+-K+交换增强,使尿排钾增多。
(二)输注大量葡萄糖液:胰岛素是餐后血钾调节的重要因素,血钾升高刺激胰岛细胞释放胰岛素,胰岛素则通过刺激Na+-K+ATP酶,促进钾进入细胞(如肌肉细胞等)。因此大量输注葡萄糖液,特别是同时应用胰岛素时可能导致低钾血症。
(三) 周期性瘫痪:
甲状腺功能亢进引起的周期性麻痹与Na+-K+ATP酶过度激活有关。原发性低钾型周期性麻痹(HOKPP)是一种较为罕见的常染色体显性遗传病,发病率约1/10万,男性多于女性。HOKPP分为家族性(FHOKPP)和散发性(SHOKPP)两型,家族性常见于西方国家,散发性则多见于亚洲国家。散发性与家族性临床特点相似,但前者发病年龄稍小,而后者无论从发病程度和发作频次上都要较散发性严重。
HOKPP多在儿童或青年时期发病,表现为四肢迟缓性瘫痪,瘫痪程度较对称,多位于肢体近端,下肢多重于上肢,偶可有肢体酸痛及麻木等主观感觉异常,严重时会引起呼吸困难,甚至恶性心律失常。本病常反复发作,发病前常有一定的诱因,如情绪激动、饱餐、剧烈运动、感冒腹泻病史、注射胰岛素及大量静点葡萄糖、月经、使用肾上腺素、糖皮质激素等。
HOKPP | 突变基因 | 机制 |
HOKPP1 | CACNA1S基因突变 | 钙通道开放异常,释放钙离子减少,最终导致钠通道的失活 |
HOKPP2 | SCN4A相关基因突变 | 钠通道异常 |
HOKPP3 | KCNE3基因相关 | 钾通道异常,钾外向电流减少,易去极化,导致钠、钙通道开放异常,导致肌无力 |
KCNE3基因定位于llq13—14,编码多肽MiRP2(MinK相关多肽2),与亚单位Kv3.4组成MiRP2-Kv3.4复合体。认为该复合体通过减少动作电位的外向电流,使静息电位不稳定,易产生去极化,导致肌无力的发生。
(四) 其它:急性应激状态致肾上腺素分泌增多,可促进钾进入细胞内;棉籽油和氯化钡中毒时,细胞膜上的Na+-K+-ATP酶持续活化,细胞外液中的钾进入细胞,而钾从细胞内流出的孔道却被特异地阻断,因而发生低钾血症;使用叶酸、维生素B12治疗贫血由于新生的红细胞利用钾增多可导致低钾血症;此外反复输入冷存洗涤过的红细胞也可引起低钾,因红细胞冷存过程中可丢失钾50%左右,输人人体后钾迅速进入细胞内造成细胞外低钾。
病例
患者四肢乏力5天入院。早晨起床时出现四肢乏力,双下肢不能抬离床面,双上肢不能举过肩。查血钾2.07mmol/L, 血钠133.55mmol/L,氯90.09mmol/L,钙0.99mmol/L。丙氨酸转氨酶225U/L,天冬氨酸氨基转移酶345.1U/L,肌酸激酶6874.2U/L,LDH615.5U/L。心电图示窦性心律,ST-T改变。入院血压正常,心率76bpm。肌张力减低,上肢肌力4级,两下肢肌力2级,膝腱反射、跟腱反射减弱。肌钙蛋白0.1μg/L,肌红蛋白1000g/L,尿蛋白(++),尿隐血(++),甲状腺功能正常。
问题 诊断?
低血钾周期性麻痹。
问题 导致肌酸激酶升高?
不少低钾血症患者可出现肌酸激酶升高,易误诊为急性冠脉综合征。其升高原因可能与低钾导致细胞膜通透性增加导致肌酸激酶外溢所致。有人认为,由于低钾血症导致肌肉代谢紊乱,游离脂肪酸在细胞内积累,影响通透性。也有认为周期性麻痹时钙通道位点突变后,间接改变了细胞的其他膜通道功能,导致细胞膜通透性增加。
问题HOPP1低血钾周期性麻痹的机制?
L-型钙通道由α1、α2、β、γ和δ5个亚基组成。低血钾周期性麻痹是由于编码,编码L型钙通道的a1亚基的CACNAIS基因(lq31-31)突变所致。其中a1亚基是离子通道的主要的功能单位,随膜电位的变化而发生构象变化,调节通道的开闭,是电压感受器的关键。
门控电流假说 | 钙通道假说 |
由于突变异常Na的内流,导致细胞内Na+浓度的升高,这可以刺激Na+-K+-ATP将细胞内多余的Na。移出细胞,同时把细胞外的K+移人细胞内,产生低钾血症,Na+泵活性增强可能是HOKPP发生的最终通路 |
钙通道开放异常, 钙离子释放减少, 影响骨骼肌的收缩使钠通道失活, 细胞兴奋性下降 |
CACNA1S如何导致低钾血症尚不清楚。门控电流假说认为由于α1结构改变导致形成一个独立于正常离子通道孔的附属离子通道,产生门控孔电流(IgP),IgP在静息电位时激活,去极化时关闭。静息电位时IgP,因内向的质子或离子流导致细胞膜的去极化及Na+超载,当细胞外血钾水平正常时,去极化并不明显,但是当细胞外血钾水平降至3mmol/L以下时,肌纤维去极化,产生电压依赖性离子通道失活、弛缓性肌肉麻痹、细胞内钠离子超载、肌细胞水肿等一系列低钾型周期性麻痹的特征。由于突变异常Na+的内流,导致细胞内Na+浓度的升高,这可以刺激Na+-K+-ATP将细胞内多余的Na+移出细胞,同时把细胞外的K+移人细胞内,产生低钾血症,很多学者甚至认为Na+泵活性增强是HOKPP发生的最终通路。但是患者平时不发病,可能因为机体的负反馈机制使机体内环境平衡,当诱因出现时,通过一系列级联放大反应,改变了Na一K一ATP与Na的亲和力和最大反应速度,在突变所致的细胞内Na+浓度异常升高的情况下,改变调定点,使血钾降低,从而导致低钾型周期性麻痹。
②:CACNA1S突变可直接影响L一型钙通道的a1亚基,a1亚基有骨骼肌双氢吡啶受体,而骨骼肌双氢吡啶受体可以激活钙通道使细胞内钙离子释放,从而引起骨骼肌收缩,突变后钙离子通道受到影响,出现开放异常,导致Ca++释放减少,这使得骨骼肌细胞膜的静息电位稳态以及阈值发生改变,不仅直接影响骨骼肌细胞的收缩,还可以直接或间接使钠通道失活,从而导致细胞膜不能兴奋性或兴奋性下降,出现肌无力症状。
低钾血症与代谢性碱中毒
血碳酸氢根浓度降低者: 肾小管酸中毒 血碳酸氢根浓度升高者: 呕吐、应用利尿剂、低镁血症、Bartter综合征、Gitelman综合征
|
低钾血症血压正常或低血压者,应进一步检测血碳酸氢根浓度。肾小管酸中毒为4种类型,其中I型(远端肾小管酸中毒)和Ⅱ型(近端肾小管酸中毒)患者常表现出低血钾、代谢性酸中毒、碱性尿,肾功能正常,血浆肾素活性及醛固酮水平均正常。高钙、急性白血病、棉酚中毒、过量使用青霉素钠盐等也常伴有低血钾,应注意鉴别和排除。
醛固酮的作用机制及分泌调节机制?
醛固酮减少 假性醛固酮减少(Gordon综合征II,I)
醛固酮增多 原发性醛固酮增多症 继发性醛固酮增多 非醛固酮导致的容量增加 Liddle综合征(假性醛固酮增多)
|
醛固酮是肾上腺球状带分泌和调节钾分泌的重要激素,醛固酮可以增加远曲小管和连接小管的钠氯协同转运子,以及位于连接小管和皮质集合管上皮钠通道(ENaC)对钠离子的重吸收,进而增加钾的分泌。醛固酮也可以增加肾脏钾通道活性,促进钾的分泌,对维持血钾、钠平衡及正常细胞外液量起重要作用。
醛固酮进入ASDN后,与胞浆受体结合形成激素-受体复合物,进入细胞核与细胞核受体结合,胞浆受体分离,最终上调醛固酮诱导蛋白表达,促进钠泵转运、生物氧化、增强管腔膜对钠的通透性来加强钠离子的主动重吸收,同时氯离子及水重吸收也随之增加,导致细胞外液量增加。
促进醛固酮分泌: 1. 动脉血压下降、循环血量减少、小管液钠浓度降低,激活RASS,促进醛固酮的合成与分泌 2. 血钾升高或血钠降低直接刺激肾上腺分泌 3. 循环血量增多,心房容量感受器兴奋,引起心房肌合成和释放心钠素,抑制醛固酮分泌及释放
|
醛固酮增多
某些疾病可引起醛固酮原发性或继发性的增多,如原发性醛固酮增多症(原醛症)、Bartter综合征、肾素瘤、肾动脉狭窄等,增多的醛固酮促进对钠的重吸收而减少对钾的重吸收,使尿钾排出增多。类醛固酮样物质产生或摄人增多:11-β羟化酶缺乏症和17-β羟化酶缺乏症产生去氧皮质酮增多,中药甘草摄入增多,均可出现类醛固酮增多症的表现。糖皮质激素产生增多:因为糖皮质激素也具有弱盐皮质激素活性,分泌增加引起低钾。Cushing综合征或异位促肾上腺皮质激素(ACTH)分泌综合征。此外,创伤、手术、感染、缺氧时由于应激,肾上腺糖皮质激素分泌亢进,尿钾排出也会增多。
Liddle综合征-ENaC获得性突变
Liddle综合征(假性醛固酮增多症)1963年Liddle等首次报道,患者表现有严重高血压、低钾血症和代谢性碱中毒,以及类似醛同酮增多样表现。位于集合管的钠通道(ENaC)对Na重新进入血管内起重要的调控作用,Liddle综合征是由于ENaC持续激活而过多的重吸收Na从而导致血容量扩张和高血压,而过多的血容量抑制了肾素一醛同酮的分泌,管腔负电位增加,同时也促进了该段K的分泌,H排出增加,从而导致高血钠、高血容量、低血钾及碱中毒。该病为常染色体显性遗传,由SCNNIB及SCNN1G基因突变所致,该基因位于16p13一p12,编码蛋向位于肾集合管细胞胞质中ENaC的B或亚单位。表现为高血压、低血钾、代谢性碱中毒、低肾素活性、低醛同酮血症。治疗包括限制盐的摄人,纠正低钾血症,钠通道阻滞剂氨苯喋啶或阿米洛利可改善其症状,而安体舒通无效。
患者男23岁,主诉发作性头痛、头晕3年入院。3年前因发热就诊发现血压180/100mmHg,药物控制不佳。2个月前测血压230/130,血钾2.94。卧位
PRA0.41ng/ml.h(正常0.93-6.56ng/ml.h),ALD20.8ng/dl(6.5-29.6ng/dl)。MRI示多发性腔隙性脑梗死。予以四种降压药物(坎地沙坦、安体舒通、比索洛尔及硝苯地平),血压仍然高。1月前复查,血钾仍然低,尿钾48.14mmol/24h。家族中奶奶及大伯死于脑出血,父亲(35)及姑姑(38)均有高血压。
问题1根据以上资料,你考虑诊断是什么?
患者有高血压伴低钾血症,尿钾>30mmol/24h,ARR为50.7,考虑原发性醛固酮增多症。但ALD不升高,不排除醛固酮假性增高。而且使用安体舒通效果不佳,早期出现脑血管疾病,而且有高血压、脑血管疾病家族史。鉴于ARR的筛查影响因素很多,易出现假阳性,仅凭临床症状及生化检查不足以排除Liddle综合征,GRA,此时需要药物试验性治疗或做基因检测明确诊断。
问题2本病采用氨苯蝶啶治疗,一周后血压稳定达标,氨苯蝶啶为何有效?
考虑本病为Liddle综合征,此病是由于ENaC过度激活所致。氨苯蝶啶及阿米洛利是ENaC的阻断剂,对Liddle综合征有一定的治疗效果。
问题3低肾素活性,为何醛固酮可以正常,甚至升高?
由于容量增加抑制肾小球旁器合成和释放肾素,导致肾素减少,相应地导致血管紧张素生成减少。低钾和高血容量均可抑制球状带分泌醛固酮,从而导致低肾素及低醛固酮。但有些患者在疾病早期可以正常,甚至增高。
问题4Liddle综合征需要与哪些疾病鉴别?
Liddle综合征常染色体显性遗传病,由于ENaC基因突变导致ENaC降解异常或者活性异常增加所致。青少年发病,无症状到成人发病。低肾素活性、低醛固酮、代谢性碱中毒、低血钾伴高血压。安体舒通无效,氨苯蝶啶或阿米洛利有效。
基因检查是诊断的金标准。
根据家族史及脑血管病并发症、发病年龄、低肾素高血压,尽管醛固酮不低,基因检测发现SCNNIB基因C1696T突变,证实本病为Liddle综合征。
本病需要与原发性醛固酮增多症(PA)、GRA、AME及肾上腺皮质增生症鉴别。对于ALD不低的Liddle综合征,很容易误诊为PA,临床症状及生化检查难以区分,药物治疗性试验及基因检查就是很好的方法。在确诊PA时,应常规排除Liddle综合征。
肾素活性降低: 1.醛固酮增多 原发性醛固酮增多症 双侧肾上腺皮质激素或肾上腺皮质腺瘤 2.醛固酮减低非醛固酮导致容量增多 Liddle综合征 柯兴综合征 21-羟化酶缺乏症 17-羟化酶缺乏症 糖皮质激素可治疗的高血压 AME 使用甘草
|
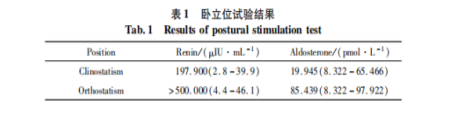
高血压患者停药物四天后行卧立位试验,血钾正常
体位 | 肾素 ng/L.h | 血管紧张素II ng/L | 醛固酮pmol/L
|
卧位 | 0.02(0.07-1.51) | 16.68(15.0-97.0) | 3200(1622-4819)
|
立位 | 0.09(0.33-3.15) | 118.5(19.00-115) | 2881(1884-8199)
|
结果提示低肾素,醛固酮正常。有些疾病在早期可表现血钾正常,醛固酮正常,给诊断带来困难。此时可以作基因检测加以排除。
高肾素、高醛固酮见于肾动脉狭窄、肾素瘤、BS等。
问题 5 Liddle综合征导致高血压的机制及临床表现解释
ENaC活性增加导致钠重吸收增加,钾外流增加,氢离子重吸收增加,随钠进入细胞内,导致细胞外液容量增加,细胞外液碱中毒,过多的细胞外液导致内源性的Endoxin增多,引起血容量增加导致血压增高。由于钾离子减低,导致一氧化氮合成减少,血管平滑肌收缩,血压升高,低钾血症进一步使内皮及肾小管受损,导致血压进一步升高。由于钠重吸收增加,抑制RAAS,导致肾素及醛固酮正常或减少。表现为低肾素高血压,醛固酮正常或降低。
糖皮质激素可抑制的醛固酮增多症:常染色体显性遗传,起病早,严重高血压,编码11-β羟化酶的CYB11B1基因和编码醛固酮合成酶的CYB11B2基因发生非对等交换,CYB11B1基因中的ACTH反应调节元件与CYB11B2基因编码区的上游启动子结合,导致醛固酮合成酶在束状带的异位表达,并受ACTH调节,醛固酮对ACTH的刺激反应强于对肾素-血管紧张素II的反应。糖皮质激素能抑制醛固酮的过量分泌,且能维持抑制效应。18-羟皮质醇和18-氧皮质醇增多,这与Liddle综合征鉴别。
AME 表征性盐皮质激素增多症为常染色体隐性遗传,由于11-β羟类固醇脱氢酶被抑制所致,导致循环中糖皮质激素可以盐皮质激素受体结合,产生盐皮质激素类样作用,尿中皮质醇代谢物/皮质酮代谢物比值明显升高。也见于儿童,表现为高血压、低血钾和低肾素、低醛固酮活性,低盐或螺内酯治疗有效,但氢化可的松或ACTH治疗加重。
关于原发性醛固酮增多症
患病率低估,原发性醛固酮增多症不仅仅是高血压低血钾,多数医生对此病缺乏认识,对原发性醛固酮增多症的筛查不清楚。值得注意的是,部分原发性醛固酮增多症患者,血钾正常;有些醛固酮正常,仅仅表现低钾血症。更为少见两者均正常的病人,这增加诊断的难度,主要依靠其他方法确诊。ARR是目前主要的筛查指标,即使血钾正常,也可以发现异常患者。

还有更大的健康问题: 心脑血管并发症 糖代谢异常 骨质疏松 抑郁症 Takotsubo综合征
|
有以下情况,需进行原发性醛固酮增多症筛查
1.持续性血压>160/100mmHg、难治性高血压(联合使用3种降压药物,其中包括利尿剂,血压>140/90mmHg;联合使用4种及以上降压药物,血压<140/90mmHg)。 2.高血压合并自发性或利尿剂所致的低钾血症。 3.高血压合并肾上腺意外瘤。 4.早发性高血压家族史或早发(<40岁)脑血管 意外家族史的高血压患者。 5.原醛症患者中存在高血压的一级亲属 6.高血压合并阻塞性呼吸睡眠暂停
|
在做检查之前,如低钾血症需要补钾,因为低钾血症可以抑制醛固酮的分泌,导致醛固酮不高(与继发性醛固酮相比),维持正常钠盐摄人。
停用对ARR影响较大药物至少4周:包括醛固酮受体拮抗剂(安体舒通、依普利酮)、保钾利尿剂(阿米洛利、氨苯喋啶)、排钾利尿剂(氢氯噻嗪、呋塞米)及甘草提炼物。
血管紧张素转换酶抑制剂(ACEI)、血管紧张素受体拈抗剂(ARB)、钙离子拮抗剂(CCB)类等药物可升高肾素活性,降低醛固酮,导致ARR假阴性,因此ARR阴性不能排除原醛症。需停用上述药至少2周再次进行检测;但如服药时,肾素活性<1ng/ml.h或低于正常检测下限同时合并ARR升高,考虑原醛症可能大.可维
持原有药物治疗。
由于β受体阻滞剂、中枢α2受体阻滞剂(可乐定或甲基多巴)、非甾体类抗炎药等可降低肾素活性,导致ARR假阳性,建议停用至少2周,如患者因冠心病或心律失常等原因长期服用B受体阻滞剂,临床医师根据患者情况决定是否停药。
如血压控制不佳,建议使用α受体阻滞剂及非二氢吡啶类CCB。
口服避孕药及人工激素替代治疗可能会降低直接肾素浓度(DRC),一般无需停服避孕药物,除非有更好更安全的避孕措施。
筛查质量影响因素
(1) 年龄:年龄>65岁,肾素较醛固酮降低明显,以致ARR升高。
(2) 性别:女性月经前期及排卵期ARR较同年龄男性高.特别对于黄体期的女性患者,如用DRC检测可能导致ARR假阳性。
(3) 采血时间、最近饮食情况、体位等。
(4) 药物因素。
(5) 采血方法。起床后2小时,采血时不要溶血,室温保存,不要放在冰上。
(6) 血钾水平。
(7) 肌酐水平
、
DRC及醛固酮检测单位各不相同,因此在统计数据时,必须根据不同单位进行换算。
醛固酮常用单位ng/dl 30 | 醛固酮常用单位pmol/L 750
| PRAng/ml·h | DRCmU/L |
ng/dl
1ng/dl=27.7pmol/L 1ng/dl=10pg/m1 | 1ng/ml·h=12.8pmol/L·min | 1ng/ml·h=8.2mU/L | |
当ARR异常,需要做确诊试验:
四种方法,比较简单就是卡托普利试验,但可能有假阳性。部分特发性醛固酮增多症可能受卡托普利的抑制。一旦确诊,需要进行分型,是临床的难点。影响到临床治疗方法的选择。
1. 影像学
2. 生化检查
3. 双侧肾上腺静脉采血(AVS)在基层医院难开展
有些需要基因分型(家族性醛固酮增多症)
高血压并低钾血症
病例2
患者男19岁,高血压10年,发现低血钾2月入院。血压200-150/130-90,期间服用厄贝沙坦、非洛地平,血压控制不佳。2个月前头痛、头晕,四肢无力,血钾2.7mmol/L尿钾66.61mmol。考虑醛固酮增多症,予以安体舒通40mg,日2次,血钾上升至3.9,血压仍然高160/110。肾上腺CT未见异常。
问题1高血压并低钾血症的原因有哪些?
高血压患者,有低钾血症。尿钾明显增高,低钾血症与尿路排钾增多有关,需排除肾小管疾病。
肾性失钾常伴随肾素一血管紧张素.醛固酮系统(RAAS)改变,可伴有或不伴有高血压。尽管高血压不是低钾血症病因诊断的决定因素,但临床常根
据是否伴有高血压分为低钾血症合并高血压和低钾血症不伴高血压。伴有高血压的低钾血症又常根据肾素醛固酮水平鉴别肾性失钾的原因。根据肾素活性和醛固酮不同水平的组合又分为3类。
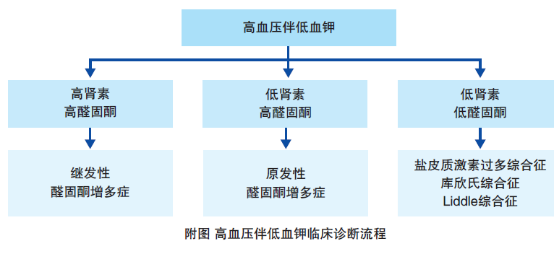
(1) 低肾素高醛固酮:
见于原醛症,由于肾上腺皮质分泌过多醛固酮,机体潴钠排钾增多,血容量增加,临床上表现为高血压,部分患者出现低血钾,肾素一血管紧张素系统受抑。血浆醛固酮浓度升高、血浆肾素活性降低,当血浆醛固酮(ng/dL)/血浆肾素活性(ng/mL·h)比值常>20,高度提示原醛症的可能。
(2) 高肾素高醛固酮:此类患者是因肾素增多引起的继发性醛固酮增多症,见于嗜铬细胞瘤、肾动脉狭窄、肾实质疾病、肾素瘤等。肾素瘤常见于青年人,表现为严重的高血压和低血钾,血浆肾素活性和醛固酮水平均升高,常规降压方案效果不好,肿瘤切除后血压和血钾恢复正常。各种原因所致肾脏缺血如恶性高血压、肾动脉狭窄及肾萎缩等可导致。肾脏供血不足,血浆肾素活性和醛固酮水平均升高。患者常有严重高血压,部分表现出低血钾,常有氮质血症或尿毒症。
(3) 低肾素低醛固酮:此类患者因非醛固酮肾上腺皮质激素或类醛固酮作用的物质增多或醛固酮受体基因突变,引起高血压和低血钾,而肾素和醛固酮受抑制分泌减少。
与高血压有关的主要有以下疾病
肾素活性升高:继发性醛固酮增多 肾性高血压 肾血管性高血压(肾动脉狭窄) 肾素瘤 肾素活性降低: 醛固酮增多;原发性醛固酮增多症 醛固酮减低非醛固酮导致容量增多 Liddle综合征 柯兴综合征 21-羟化酶缺乏症 17-羟化酶缺乏症 糖皮质激素可治疗的高血压 AME 使用甘草 肾素活性正常,醛固酮正常 髓质海绵肾
|
问题2 低肾素低醛固酮有哪些?
Liddle综合征可引起高血压、低血钾、低肾素及低醛固酮。由阿米洛利敏感性上皮钠通道(ENaC)的p或亚基突变导致的常染色体显性遗传疾病。因远端肾小管EnaC过度激活,肾小管病变重吸收Na和分泌K亢进,钠不适当重吸收增加,钾排泄增加,导致容量性高血压和低血钾,与原醛症相似,但血浆醛固酮和肾素水平低下,盐皮质激素受体拮抗药螺内酯对其无效,ENaC阻断剂氨苯蝶啶有效,因此,又称为假性醛固酮增多症。
肾上腺皮质其他激素分泌异常所致高血压低血钾
先天性肾上腺增生症(CAH):本病是由于皮质激素合成过程中所需酶的先天性缺陷而引起的一组疾病,为常染色体隐性遗传病。如11α或11β-羟化酶缺乏症(11-去氧皮质酮或11-去氧皮质醇分泌增多)导致储钠排钾,故产生高血压、高血钠和低血钾,皮质醇和醛固酮减少,ACTH代偿性分泌增加。同时存在性激素合成异常引起表型性征异常,对本症有提示作用。因皮质醇和醛固酮减少,出现皮肤黏膜色素沉着,女性出现多毛,使用糖皮质激素可使症状好转。
17α-羟化酶缺乏症(11-去氧皮质酮和皮质酮分泌过多)出现高血压及低血钾和代谢性碱中毒,并有原发性闭经。
表象性盐皮质激素过多综合征:为先天性11B一羟类固醇脱氢酶缺陷,皮质醇不能转变为皮质素,增多的皮质醇激活盐皮质激素受体,导致水钠储溜,表
现为低肾素性高血压、低血钾性碱中毒及尿17一羟及游离皮质醇排出量减低。24h尿皮质醇(或代谢产物)/皮质素可用于筛查该病。
Geller综合征:又称妊娠加重型高血压。因编码盐皮质激素受体(MR)的基因发生突变引起的常染色体显性遗传病。MR的活性增加,使钠吸收增加。此类突变携带者在非妊娠期也会发生高血压,但妊娠期会显著加重,可能孕后孕酮增加,进一步活化MR的结果。多于20岁前发病,血浆肾素活性和醛固酮水平低,血清钾水平降低或正常,很少发生蛋白尿、水肿及神经系统症状,可区别于妊娠高血压综合征。
皮质醇增多症:为多种病因造成肾上腺皮质分泌过多的糖皮质激素所致。表现为高血压和低血钾性碱中毒,伴有向心性肥胖、满月面、多血质、紫纹及痤疮;血皮质醇浓度升高,无昼夜节律,小剂量地塞米松抑制试验不被抑制。肾上腺病变以双侧增生最为多见,也可为腺瘤或癌。
问题3进一步需要做哪些检查来确诊?
尿常规、肾功能及肾脏超声检查了解有无肾脏疾病导致的高血压。
肾素活性,醛固酮,血管紧张素II及卡托普利试验;血气分析有无酸碱失衡;肾上腺皮质激素功能检查。
醛固酮升高,卡托普利试验不受限制
PRA(ng/ml.h) AT-II(pg/ml) Ald(ng/dl) 前0.035 0.18 16.66 后0.065 2.29 18.76 |
血钾:2.8,血气分析:pH7.448,SB 31.0mmol/L。
ACTH34.7(pg/ml),血皮质醇11.48μg/dl,24小时UFC61.18μg,小剂量地塞米松抑制试验:血皮质醇0.33μg/dl,17-OHP 0.5ng/ml,DHEAS250μg/dl。CT示左侧肾上腺结合部饱满。
问题3根据以上资料你考虑什么?
患者有低钾血症伴碱中毒;醛固酮增高,不受卡托普利抑制,肾素活性明显减低,自主分泌或合成增多。肾上腺CT未见异常,排除肾上腺肿瘤。考虑原发性醛固酮增多症,但患者发病年龄早,不符合特发性醛固酮增多症,考虑家族性醛固酮增多症,共有四型,最常见为GRA,即FH-I。
临床表现:高血压、低血钾、低肾素、高醛固酮水平。小剂量糖皮质类固醇反馈性抑制ACTH,从而抑制ACTH调控的醛固酮合成。
提示GRA诊断的临床线索:早发高血压家族史和(或)早发脑中风家族史。实验室检查:血与尿的生化表现支持原发性醛固酮增多症的诊断,如醛固酮/肾素比率升高,醛固酮水平增加,血浆肾素活性降低(纠正血钾以后测定),合并低血钾或血钾不低。尿中18-酮与18-羟皮质醇升高。
GRA诊断的金标准:Southern印迹法及(或)长片段聚合酶链式反应(long-rangePCR),确定存在嵌合基因。
GRA可出现不同程度的高血压,有时容易被误诊为原发性醛固酮增多症,但却在发病早期有较高致死率。有以下表现者应考虑该病:20岁以前患高血压病,高血压家族史,50岁前出现脑出血,难治性高血压和低血钾。Southern印迹法或长距离聚合酶链反应(PCR)法检测CYP11B1/CYP11B2的嵌合基因可明确诊断。
GRA的处理:一旦确诊,即刻给予小剂量糖皮质激素,如地塞米松0.125~0.250mg/d,或强的松2.5~5.0mg/d,降低醛固酮产量。小剂量非常重要,
维持昼夜皮质醇调节;完全抑制ACTH-调控的嵌合11β-羟化酶-醛固酮合成酶基因产生醛固酮;一般3~5d后血压便可下降。严重病例:地塞米松+盐皮质类固醇受体拮抗剂(安体舒通或依普利酮),也可以联合使用其他降压药。因为激素有副反应,儿童GRA,可给予依普利酮。
拓展:GRA发生的机理
生理情况下,醛固酮合成酶基因(CYP11B2)在肾上腺皮质球状带表达,受血管紧张素Ⅱ调控作用合成醛固酮,11β羟化酶基因(CYP11B1)在束状带表达,受ACTH调控。GRA者CYP11B1的调控序列与CYP11B2编码序列融合形成一个新的“融合基因”,在束状带表达,不受血管紧张素Ⅱ和血钾调控,而受ACTH调控,具有醛固酮合成酶编码11β-羟化酶的基因CYP11B1(合成糖皮质激素)与编码醛固酮合成酶的基因CYP11B2(合成盐皮质类固醇,如醛固酮)嵌合突变,即醛固酮合成酶基因编码序列与糖皮质激素合成酶基因的调控序列交叉重组,醛固酮的合成受促肾上腺皮质激素(ACTH)的调控,导致ACTH-依赖性醛固酮合成亢进,并引起1I羟皮质醇及18-酮皮质醇合成增多。不受血管紧张素II的调节,醛固酮分泌可以被糖皮质激素所抑制。
Bartter综合征
常染色体隐性遗传性肾小管疾病为肾脏髓袢升支粗段和远端集合管离子通道的基因突变所致的常染色体隐性遗
传代谢性疾病,各年龄段均可发病,常于婴儿及儿童时起病,50%在5岁以前发病,临床罕见。根据不同的基因突变,将其分为5(I~V)型:I型致病基因为
SCL12A1,位于15q15~21;Ⅱ型致病基因为KCNJ1,位于11q24~25;III型致病基因为CLCNKB,位于1q36;IV型致病基因为BSND,位于1p31;V型致病基因为CASR,位于3q13~21。临床主要表现为低血钾、低血钠、低血氯、代谢性碱中毒、尿钾及尿氯排出增多,血浆肾素活性及醛固酮水平增高,但无高血压。成人Bartter综合征,临床症状较轻,醛固酮可以正常。肾脏活检提示肾小球球旁器明显增生,电镜下小球旁器细胞内分泌颗粒增多
基本病变为髓襻升支后段Na-K-2Cl转运体异常致使NaCl重吸收障碍。远小管中Na增高,Na-K和Na-H交换均增强,钾排泄增多,则产生低血钾代谢性碱中毒,尿呈碱性;并刺激RAS分泌增多,出现高肾素、血管紧张素及醛固酮增血症,由于PGE及激肽分泌也增多,故不发生钠潴留和高血压,在低血钾的同时尿镁和尿钙排泄也增多,故发生低血镁及低血钙所致的肌抽搐或肌痉挛。
尿浓缩力减低,后期由于长期低血钾可发生失钾性肾病及肾性尿崩症,应用消炎痛治疗往往有效。在诊断前应注意有无慢性频发呕吐、腹泻或神经性厌食等,这些情况可发生与Bartter综合征相同表现,故称为假性Bartter综合征(无原发性肾小管异常),在这些情况下均可发生低血钾和酸碱平衡障碍,故应注意鉴别。
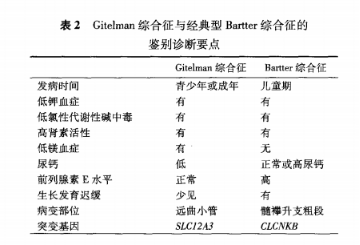
Gitelman综合征:为常染色体隐性遗传,位于远曲小管对噻嗪类利尿剂敏感的转运子基因失功能型突变。罕见,易漏诊及误诊。
NCCT参与肾小球滤过液中5%-10%氯离子及钠离子的重吸收。当NCCT缺陷时,导致重吸收减少,RAAS激活,导致容量减少,排钾增加,导致低血钾。编码CIC-Kb的氯通道CLCNK8基因突变(BartterIII)及编码肝转录因子1-b(HNF1B)基因突变也可产生类似症状。低镁血症<0.7,低尿钙(尿钙肌酐比<0.2)应怀疑NCC功能障碍。行50mg氢氯噻嗪(完全阻断NCC)试验观察使用药物前后排泄分数的变化程度,正常FECL<2.86%。尿钾>25mmol/24h
至今GS的确切发病率尚不清楚。青少年或成年期起病,临床表现差异极大。
GS患者一般在青少年或成年时发病,多于6岁以后起病,血尿生化异常可早于临床症状出现。临床上表现为低镁和低尿钙与Bartter综合征相区别。镁正常,钙不低,也不能排除。
主要表现为低钾血症及低镁血症,有些无症状或仅表现乏力、纳差,严重会出现四肢抽搐、瘫痪、痛性痉挛、晕厥和横纹肌溶解继发急性肾损伤,有些出现室性心律失常、心脏骤停。
多数患者临床表现为乏力、疲劳、口渴、多尿等非特异性症状,治疗后预后良好,因此GS曾被认为是一种良性肾小管疾病。但近年的研究显示,GS患者的生活质量明显下降,少数患者甚至可出现生长发育迟缓、软骨钙化、横纹肌溶解和室性心律失常等严重临床症状。此外,长期的低钾低镁可能导致糖代谢异常、肾功能受损等并发症而影响预后,因而早期诊断、合理治疗及监测病情非常必要。
肾性丢钾导致的低钾血症,伴有MB,RAAS激活(高肾素),但血压正常
问题1钠氯协同转运体(NCC)的结构与功能?
生理情况下,通道蛋白NCC位于肾脏远曲小管上皮细胞的管腔侧,参与肾小球滤过液中5%~10%氯离子和钠离子的重吸收,是机体维持水、电解质平衡的一道重要防线。
问题2Gitelman综合征的临床特点?
Gitelman综合征是最常见的遗传性肾小管疾病之一,患病率为1/40000~1/4000。Gitelman综合征是由编码噻嗪类利尿剂敏感的钠氯协同转运体(NCC)的SLC12A3基因突变导致NCC结构和(或)功能障碍,氯离子和钠离子从远端肾小管重吸收减少,肾脏重吸收水减少,继发性RAAS活化、肾性失钾和钙
重吸收减少。
常于青少年或成年早期起病,轻型患者可无症状或表现为轻度乏力和纳差;严重患者会出现四肢抽搐、软瘫、痛性痉挛、晕厥和横纹肌溶解继发急性肾损伤,甚至因为严重室性心律失常导致心搏骤停。
临床特征
1.低血钾:引起的相关症状,从无到严重并发症,其中血钾小于3.5mmol/L时,24小时尿钾大于25mmol
2.低血镁<0.7mmol/L)
3.血压正常或低血压
4.(成人尿钙肌酐比<0.2mmol/mmol)
5.代谢性碱中毒
6.raas活化
临床表现
1.全身症状疲乏、口渴、多饮、嗜盐。
2.神经-肌肉系统肌无力、痛性痉挛、抽搐、惊厥发作、肢体麻木、感觉异常、横纹肌溶解、瘫痪、头晕、眩晕、共济失调和假性脑瘤
等。
3.心血管系统心悸、晕厥、血压正常或偏低和室性心律失常等。
4.消化系统便秘、呕吐等。
5.泌尿系统多尿、夜尿增多、遗尿、蛋白尿和肾功能不全。
6.骨关节系统关节痛、软骨钙质沉着症。
7.内分泌和生长发育生长迟缓、青春期延迟;长期低钾和低镁的患者糖尿病或者糖耐量减低的比例并不少见。
8.眼部症状少数患者会出现视物模糊和巩膜脉络膜钙化。
问题肾脏表现?
Gitelman综合征患者蛋白尿和肾功能损害的原因主要与长期低血钾相关,后者可导致肾小管间质损伤和囊肿形成,蛋白尿以小管来源为主;其次因RAAS长期激活,可能直接或间接导致肾小球节段硬化,极少数病例合并有其他肾小球疾病,如IgA肾病、C1q肾病等,但其可符合肾性失钾。
肾脏超声肾脏形态多正常,长期低钾患者可出现肾囊肿,可用于排除其他因为肾脏结构异常导致的肾性失钾。
氢氯噻嗪试验通过小剂量氢氯噻嗪(50mg)直接阻断NCC,观察使用前后氯离子排泄分数的变化程度(△FECl),与正常对照进行比较,评估NCC功能。中国人群中△FECl<2.86%时诊断Gitelman综合征的灵敏度和特异度分别为95.7%和95.8%。目前临床采用改良的氢氯噻嗪试验较为安全且简便易行,成本低,但对于怀疑Bartter综合征的患者,需注意监测血钾进一步降低的风险。
。
。
问题鉴别诊断
确诊需要基因诊断
基因检测是诊断Gitelman综合征的金标准。检测到SLC12A3纯合突变或复合杂合突变可确诊,单杂合突变的患者需结合临床,新发现的突变需要体外功能试验确定突变的致病性。
慢性肾性失钾导致的低钾血症,伴有代谢性碱中毒,血浆肾素-血管紧张素-醛固酮系统活化(立位),但血压正常或偏低的患者,应考虑失盐性肾病。
应高度怀疑NCC功能障碍。进一步的氯离子清除试验(氢氯噻嗪试验)有助于定位NCC功能障碍,并能初步判断损害程度。需要注意的是,即使血镁正常或者尿钙不低,并不能除外Gitelman综合征。
需要除外长期服用氢氯噻嗪类利尿剂或缓泻剂,其他因为药物、免疫病(如干燥综合征)或者单克隆免疫球蛋白病等导致的远端小管功能障碍。
Gitelman综合征主要与以下两类疾病鉴别:
1.其他原因的低钾血症首先需要根据病史和24小时尿钾与血钾水平比较,确定是否肾性失钾。然后检测尿氯水平,如尿氯排泄水平不高
(<20mmol/L)则需警惕呕吐、腹泻等情况,反之则考虑存在肾性失氯;合并高血压应警惕原发性/继发性醛固酮增多症,库欣综合征;如
合并代谢性酸中毒要警惕肾小管酸中毒。其次除外其他药物(特别是利尿剂和中药)、免疫病和浆细胞病所继发的肾性失钾,可以通过相应的
检查帮助鉴别。
2.其他失盐性肾病如Bartter综合征。经典Bartter综合征(Bartter综合征Ⅲ型)由编码氯离子通道ClC-Kb的CLCNKB基因突变所致,其
起病相对较早(3岁以前),低钾程度更重,更易出现生长迟缓、多尿,患者血镁水平多正常,尿钙水平正常或偏高。患者对氢氯噻嗪试验有
反应,说明NCC功能正常;但呋塞米试验没有反应,有助于临床鉴别。基因检测有助于确诊。
问题如何治疗?
Gitelman综合征以对症治疗、电解质替代治疗为主,以期达到缓解症状、提高生活质量、避免严重并发症的目标。总体治疗原则如下:
1.替代治疗推荐高盐饮食,进食富含钾、镁的食物,口服氯化钾、门冬氨酸钾镁、硫酸镁和氯化镁等药物,紧急或严重情况下可静脉输注
钾盐和镁盐。2017年改善全球肾脏病预后组织(KDIGO)专家争议共识建议血钾和血镁治疗目标分别为3.0mmol/L和0.6mmol/L。
2.其他治疗保钾利尿剂(如螺内酯、依普利酮)、肾素-血管紧张素系统抑制剂(低血压时慎用)抑制RAAS活化,前列腺素合成酶抑制剂(吲
哚美辛等)有助于减少补钾药物的剂量,改善低钾相关症状。但需注意监测相关药物副作用。
3.患者管理和宣教强调个体化的疾病管理,培养和加强患者自我监测症状体征,按时使用药物、适时就医、规律随诊,并需要重视患者的
心理健康。
4.特殊情况对于妊娠期、围手术期及合并其他疾病的Gitelman综合征患者,应加强监测并积极随访,及时调整药物,避免病情加重及严
重并发症。
19.Gitelman综合征(GS)-NCT
罕见的常染色体隐性遗传性肾小管病,其特征为低血钾代碱伴低血镁及低钙尿(BS为高钙尿),伴有高肾素血症及高血管紧张素Ⅰ血症,而血管紧张素Ⅱ及醛固酮不增高(ACE正常,可能是糜蛋白酶活性缺乏),其病因是远小管噻嗪-敏感性NaCl转运体(cotransporter)基因译码突变所致。证明其Na-Cl转运体基因编码SLC12A3突变所致。本征持续过久,可发生软骨钙(焦磷酸钙)沉着病,骨镁排空,反复发作的关节痛及积液,又称假性痛风。
病例
患者男14岁,因胸闷、气短1周,四肢乏力5天入院。1周前于当地医院查血钾低2.59-2.81mmol/L诊断为低钾血症。予以补钾后血钾为3.47mmol/L。为了进一步诊疗收入院。其祖母有低钾血症。血压112/82,两下肢无浮肿,肌力正常。
尿pH6.5 相对体积质量1.025 尿蛋白阴性 尿钾49.44mmol/24h(25-125) 尿钙2.5(25-7.5) 血糖4-6mmol/L 血钠138 血氯97.0 血钾3.17 钙2.47 镁0.66(0.70-1.1) 肌酐53.8 pH7.434 血碳酸氢根AB31.7(SB30.7) |
问题19.1根据以上资料,患者有哪些异常?
1. 低钾血症
2. 低镁血症
3. 低尿钙
4. 代谢性碱中毒
考虑Gitelman综合征。
问题19.2从卧立位试验结果发现肾素活性增高,请解释其机制。
有数百种基因突变与NCCT功能或结构异常有关。Gitelman综合征是一种常染色体隐性遗传的失盐性肾小管疾病。因1966年美国医生Gitelman等首先报道了3例家族性低钾、低镁、低尿钙及代谢性碱中毒而得名。该病曾长期被认为是Bartter综合征的一个特殊亚型,直至1996年其致病基因被成功克隆。现已明确,GS的病因是编码位于肾远曲小管的噻嗪类利尿剂敏感的钠氯共同转运体(NCCT)蛋白的基因SLC12A3发生功能缺失突变导致NCCT的结构和/或功能异常,从而引起肾脏远曲小管对钠氯重吸收障碍导致远端肾小管重吸收钠离子、氯离子减少,导致远端肾小管及集合管Na+-H+和Na+-K+交换增加,水丢失导致低血容量、肾素-血管紧张素一醛固酮系统(RAAS)激活,最终导致低血钾和代谢性碱中毒等一系列病理生理和临床表现。由于Na+重吸收减少,Na+-Ca++交换增加,管腔侧Ca++重吸收增加,导致尿钙减少。镁离子减低与Na+离子依赖的镁离子重吸收减少有关。
醛固酮常用单位ng/dl 30 | 醛固酮常用单位pmol/L750
| PRAng/ml·h | DRCmU/L |
ng/dl
1ng/dl=27.7pmol/L 1ng/dl=10pg/m1 | 1ng/ml·h=12.8pmol/L·min | 1ng/ml·h=8.2mU/L | |
问题19.3Gitelman综合征的鉴别诊断?
主要与Bartter综合征鉴别,两者均有低血钾、肾胜失钾、低氯性代谢性碱中毒、RAAS激活但血压不高。鉴别要点主要是发病年龄、是否存在低尿钙、低血镁及是否合并生长发育迟缓,基因检测可以明确。