血磷代谢紊乱及管理
血清磷浓度
| mmol/L |
正常
| 0.96-1.15 |
低磷血症
| <0.96 |
高磷血症 | >1.15 |
低磷血症分阶: 轻度0.8-0.96;中度0.3-08;重度<0.3
| |
高血磷与慢性肾脏病及血液透析的关系研究相当透彻,而低血磷症不被人们重视。低血磷并不罕见,临床上至少有0.43%的患者有严重的低血磷,一些特定的人群可能更高:酗酒、营养不良、脓毒血症及酮症酸中毒。重度的低血磷症与横纹肌溶解、溶血、呼吸衰竭及心力衰竭有关。严重的低血磷症增加死亡率。
磷是构成细胞组织,参与细胞、组织代谢的重要元素。85%磷元素储存在骨组织及牙组织,14%分布在细胞内及1%在细胞外液中。保持磷的平衡。血磷有生理节律,凌晨迅速下降,至11点达低点,后上升,16点升高至平台,到凌晨1到3点达高峰。
正常血清磷浓度为0.96-1.15mmol/L。血磷主要有机磷或磷脂(70%)及无机磷(30%)形式存在。血清无机磷10%与蛋白结合,5%为钙、镁复合物,85%为H2PO4-和HPO42-,其他形式的游离无机磷离子可以忽略不计。磷从小肠吸收,正常血磷在0.87-1.45mmol/l。血磷是通过肾小管排泄磷来实现血磷水平稳定的,尿磷的排泄基本与从小肠吸收的磷相当,血磷水平与FGF1,25-维生素D3及PTH有关。
肠道通过细胞和细胞间两条途径吸收磷,与镁离子相似。经上皮细胞的磷转运由钠依赖酶介导。磷酸盐通过肠上皮细胞刷状缘泵入细胞内,随后被转运至细胞外。腔内pH和离子梯度决定了某种离子更易于吸收。低磷饮食和1,25(OH)2维生素D3是促进肠道钠-磷协同转运蛋白(NaPi-IIb)活性的最主要因素最近研究表明NaPi-IIb活性并不依赖维生素D3。代谢性酸中毒、雌激素和表皮生长因子(EGF)均可促进NaPi-IIb表达。低磷饮食刺激肾脏1α-羟化酶活性,增加1,25(OH)2维生素D3合成。
问题 磷吸收?
主要在小肠上部,肠道通过跨细胞和细胞间两条途径吸收磷。有机磷必须转变为无机磷才能吸收。钠-磷协同转运蛋白(NaPi-IIb)主要分布在十二指肠和空肠刷状缘,参与肠道磷的吸收。饮食中的矿物质含量影响磷吸收,钠钾促进磷吸收,而镁则减少磷吸收,摄入大量钙也会影响磷的吸收。
低磷饮食和1,25(OH)2维生素D3是促进肠道钠-磷协同转运蛋白(NaPi-IIb)活性的最主要因素最近研究表明NaPi-IIb活性并不依赖维生素D3。代谢性酸中毒、雌激素和表皮生长因子(EGF)均可促进NaPi-IIb表达,从而促进磷吸收。
体内磷主要从肾脏排泄350-1300mg,粪磷排出大约500mg。肾脏磷的平衡主要通过近曲小管磷重吸收来调控。随摄入磷变化,肾脏排磷也发生相应变化。近曲小管有三种钠-磷协同转运蛋白,在NaPi-IIa完成大部分的磷重吸收。正常下,80%被近曲小管磷重吸收,低磷饮食时几乎完全吸收,高磷饮食则抑制近曲小管磷重吸收。口服镁剂,可使磷排泄减少。
钠-磷协同转运蛋白 | 分布 |
I | 在细胞顶膜 |
II 三种异构体:a,b,c
| 细胞顶膜,a,c在近曲小管 b在肠道,负责肠道的磷吸收 |
III | 基膜外侧 |
肾是主要排磷器官,通过NaPiII转运体完成。NaPiII有三种异构体:a,b,c。b在肠道表达,a,c仅在肾小管表达,PTH可以使其灭活。
问题NaPi-IIa转运磷的机制?
此转运蛋白在近曲小管上皮及肠上皮细胞顶膜上表达,受激素及维持自身稳态的代谢因子控制。上皮细胞磷转运,由II型钠-磷共转运体(NaPi-II)活性决定。
在PTH及非激素等作用下,活化细胞信号转导途径,引起NaPi-IIa内化,通过膜表面的网格蛋白形成内体(内涵体),在溶酶体降解。PTH与顶膜激活蛋白激酶的受体结合,激活PKC和cAMP-PKA途径,引起NaPi-IIa灭活的机制不清。由于NaPi-IIa数目减少,磷重吸收也减少,排泄增多,抑制磷重吸收。
胰岛素可促进近曲小管磷的重吸收,同时抑制PTH的磷酸化。GH则通过IGFI刺激NaPi-IIa转运磷。甲状腺素也可促进近曲小管磷重吸收。降钙素及糖皮质激素则抑制吸收。
正常磷代谢受多种因素调控 PTH 抑制磷重吸收,降磷(a,c) FGF23抑制磷重吸收,降磷(a,b,c) 活性维生素D具有双重性(b) 低磷血症 尿磷减少 高磷血症 尿磷增加 | 每天吸收800-1400mg 尿排出700mg 肠道排出500。
|
问题 FGF23 增加如何降低血磷水平?
由骨细胞产生,基因定位于12p13。是成纤维细胞生长因子家族中分子最大的一类蛋白,含有251个氨基酸。在血磷调节中起重要作用。
FGF23作用于NaPi-II a,c转运体及b,抑制磷的重吸收,促进尿磷酸盐的排泄。抑制25-羟基维生素D3-1α-羟化酶,上调25-羟基维生素D-24羟化酶的表达,使1,25(OH)2D3的生成减少、降解增多。抑制PTH的合成。PTH可以调节NaPi的表达从而调节肾脏重吸收磷。
问题 1,25(OH)2D3对血磷的调节作用?
1,25(OH)2D3可增加NaPi-IIb转运体的活性,使肠上皮细胞对磷的重吸收增加,同时促进肾小管上皮重吸收磷,使血磷水平升高。血磷促进PTH分泌合成,从而抑制NaPi-II a,c的活性,减少肾脏对磷的重吸收,避免血磷升高。
问题 磷有何作用?
在体内参与许多重要生理活动,骨的形成,参与ATP及体内代谢相关的各种酶,是细胞膜磷脂和核苷酸的组成成分。磷酸盐是人体重要的缓冲系统,可维持内环境酸碱平衡稳定,并调节成骨细胞的分化。
高磷血症
肾脏排泄磷障碍 肾功能不全、肢端肥大症、甲状旁腺功能减退症、假性甲状旁腺功能减退症、使用肝素及家族性肿瘤样钙质沉着症 分解代谢加快或细胞溶解 高分解状态 组织损伤(高温、挤压综合征、暴发性肝炎) 溶血性贫血 横纹肌溶解症 肿瘤溶解综合征 细胞间磷转移 代谢性酸中毒 呼吸性酸中毒 静脉输入磷制剂 磷酸盐、脂质体二性霉素B、磷苯妥英
|
肾脏排磷的能力是有限的,低磷刺激PTH及FGF的分泌。PTH抑制肾脏对磷的重吸收,而FGF23减少肾脏对磷的重吸收,促进磷的排泄,低磷性佝偻病由于肾小管对磷的重吸收障碍,导致磷的排泄增加,FGF23的过多表达或分解障碍是大部分低磷性佝偻病的主要原因,在这些疾病中,表现为FGF23水平增高。它主要作用于肾小管刷状缘膜上的IIa型钠-磷协同共转运体使其降解,还能抑制1α-羟化酶,使活性维生素D合成受阻。
急性、慢性肾功能衰竭是高磷血症的主要原因。当肾脏排泄能力降到一定程度,GFR<40ml/min时,低于食物摄入的磷时,将导致血磷升高。甲状旁腺功能减退时,由于PTH减少导致PTH抑制磷重吸收作用减少,引起血磷升高,FGF23可以抑制血磷的进一步升高。PTH只是引起血磷升高的原因之一。低PTH可导致低血钙,低血钙进一步降低磷的清除率,补充活性维生素D可以纠正低血钙从而降低血磷,即使PTH很低。由于血磷增高,磷对PTH的抑制作用减弱,导致继发性PTH。
一些非肾功能不全的疾病如影响肾小管排出磷,也会导致高磷血症,如肢端肥大症、甲状旁腺功能减退症、假性甲状旁腺功能减退症、使用肝素及家族性肿瘤样钙质沉着症。
家族性肿瘤样钙质沉着症大多无症状或轻微,严重的高磷血症往往伴有低钙血症。可能与酸中毒、高血钾及高尿钙并存。广泛的磷酸钙盐沉着会导致脏器功能异常。
家族性肿瘤样钙质沉着症是FGF23失活突变或 O-糖基转移酶 GALNT3突变(糖基化并激活 FGF23)。突变的FGF23或FGF23产物易降解,这样失活的C末端片段血液水平远高于具有活性的FGF23。临床表现:局部高度成骨,在关节周围异位钙化,如肩关节及髋关节。高血磷是由于肾TRP(非选择性阳离子通道家族)表达增加所致,血清骨化三醇也升高,PTH可能低或正常。促进肠道钙吸收。在儿童或成人发病,好发于黑人。由于骨化三醇升高及肠道钙吸收增加,尽管存在高血磷但不发生继发性甲状旁腺功能亢进。治疗较为困难。
特别在肾功能受损时,部分高磷血症是由于快速输入磷制剂或口服一些药物(脂质体二性霉素B、苯妥英钠)所致,某些病理情况下(代谢性酸中毒、呼吸性酸中毒)磷从细胞内液转移到细胞外,导致血磷升高。细胞溶解引起的高磷血症将是严重的,如肿瘤溶解综合征等。有些可能由于肠道磷负荷过多所致,如口服润便、肠道准备或催吐药等。
大多高磷血症无症状或轻微,严重的高磷血症的临床表现常与伴随的低钙血症有关,引起不溶性的磷酸钙沉着。患者出现强直、肌肉痉挛、瘫痪、抽搐,与其他代谢紊乱常共存(高钾血症、酸中毒及氮质血症)使临床表现更为复杂。全身性磷酸钙沉着会导致器官功能不全,加重肾功能衰竭。
高血磷的危害极大,高血磷导致继发性甲状旁腺功能亢进及囊性纤维骨炎。长期高血磷,磷酸钙沉积在血管壁,超过6.5mg/dlmg/dl(1mg/dl=0.0323mmol/与早期死亡率风险增加有关。
KDIGO更重视CKD血磷的控制,建议CKD3—5D期患者血磷应控制在0.81—1.45mmol/L。在CKD从3期进展到5期(非透析)的过程中,高磷血症的发生率从4.2%上升到23.4%,在一项对40538例透析患者研究中,血磷水平超过1.62mmol/L的患者比率为67%,超过1.29mmol/L为88%。上海及北京地区ESRD患者中血磷>1.78mmol/L的比率分别为56.1%和44.8%。高磷血症在CKD患者中普遍存在,控制情况尚不理想。我国血液透析磷达标率可能不到50%(3.5-5.5mg/dl,1mg/dl=0.0323mmol/),高血磷不受重视,高血磷的危害未被充分认血识。
日常只需要磷700mg,而我们日常摄入远高于此需要量。随着GFR的下降,低于30ml/min时,此时磷排泄量不足以补偿食物磷的摄入量,如果饮食磷摄入不减将导致血磷进一步升高。在CKD3期以上,要求控制在2.7-4.6mg/dl之间。当血磷高于4.6mg/dl(1.44mmol/l)时就要限制膳食中的磷,每天不超过800-1000mg/d。食物磷入量应限制在800mg/d以下(最佳入量为500mg/d),5-7mg/kg.d。
低磷的食物 蛋清 粉丝 凉粉 土豆 白薯 萝卜 芋艿 水果及蔬菜。
| 注意食物添加剂中的磷,目前的食物成分表没有含磷的指标,应咨询专业医生。
|
CKD低蛋白饮食要求以牛奶、蛋类为主。乳类、肉类及豆类是食磷的常见来源,在肠道的吸收60%以上。过度减少膳食磷可能减少蛋白质的来源,不要因为限制磷而放弃蛋白质,尽量选择低磷/蛋白质比值的食品,既满足蛋白需要,又能减低磷摄入。在CKD4期,如乳清蛋白是最好的选择。在进食牛奶时不要超过250ml(1L相当于1g磷)。在行低蛋白饮食时,首先考虑蛋白需要量,至少保证50%为高生物价蛋白质,再根据磷/蛋白选择低磷/蛋白比的食物。
植物来源的食物磷吸收低,加热后可能使吸收量增加,通过水煮减少磷摄入可能达不到目的。另外来自食物添加剂为隐藏磷,大多为无机磷,吸收利用率高,其摄入量不可忽视,估计有些超过1000mg/d。有些药物可能导致血磷增高如果糖糖二磷酸,脂质体二性霉素,不要输注磷制剂及含磷的药物(苯妥英钠、口服排便药物及催吐药)
在低蛋白饮食中,800-1000mg/d还是能容易实现的。有时可能需要使用磷结合剂,可以减少食物磷的吸收。肠道磷结合剂包括传统的磷结合剂,如含铝、钙、铁、镁的磷结合剂,以及新型磷结合剂,如盐酸(碳酸)司维拉姆和碳酸镧等。
由于含铝的磷结合剂可能引起铝中毒,含铝磷结合剂降磷效果明显,但有导致贫血、骨病和中枢神经系统损害等不良反应,现已较少使用,短期可使用。含钙磷结合剂(碳酸钙和醋酸钙等)目前用于高磷血症治疗,能有效降磷,但含钙的磷结合剂有可能导致高钙血症,特别是与活性维生素D合用时更容易发生,使患者出现血管和其他部位转移性钙化。含钙的磷结合剂应慎用,高血钙(高于10.2mg/dl)患者应避免使用。其他包括含铁、含镁磷结合剂、烟酸等,由于其结合磷效力弱或不良反应,目前均未在临床广泛使用。
盐酸司维拉姆为不含铝、不含钙、亦不含任何金属成分的多价阳离子树脂(多聚盐酸丙烯胺),在胃肠道内水合膨胀数倍于原体积的凝胶,生理pH值下其胺基几乎完全质子化,通过离子交换结合磷与胆汁酸。
pH为7时司维拉姆与磷发生高效结合,在体内其与磷的结合主要在近端小肠。该药不被胃肠道吸收,随粪便完全排泄,安全性高,不导致高钙血症。司维拉姆能有效控制CKD患者的高磷血症,其降低血磷的能力与含钙磷结合剂相当,但较少发生高钙血症、PTH过度抑制,同时减缓冠状动脉钙化进展。司维拉姆显著降低LDL-C,这一作用对糖尿病、高血压、血脂异常及合并心脏疾患的人群尤为有益。该药常见副作用有:
(1)胃肠道反应;
(2)盐酸思维拉姆的氯离子吸收入血液可引起血浆碳酸氢盐水平下降,第二代碳酸思维拉姆用碳酸根替代氯离子与多聚丙烯酰胺结合,从而在降血磷同时显著提高血清碳酸氢盐水平;
(3)大剂量的盐酸司维拉姆可引起多种脂溶性维生素如维生素D、E、K吸收减少。同时,在pH值较低或>7时,思维拉姆结合磷的能力降低。
镧是一种稀有元素。很早发现摄入镧盐会降低磷的摄入,造成血中磷酸盐浓度过低。碳酸镧是新一代不含铝和钙的磷结合剂,比传统的磷结合剂更有效、安全,可有效降低病人血清中的磷酸盐水平,而不会导致血钙升高和其他严重副作用,耐受性好。
碳酸镧在消化道中释放出3价阳离子镧,对食物中的磷具有高度的亲和力。镧与磷结合形成低水溶性的镧盐后,极少被胃肠道吸收,通过粪便排出体外。体外研究显示,镧和磷在pH为1—7条件下均能保持较高的结合力,尤其在pH为3—5时最佳,因而在胃或十二指肠和空肠均能与磷高效结合,且不影响脂溶性维生素的吸收。碳酸镧与磷的结合力高于司维拉姆,在pH为3的条件下,碳酸镧与磷的结合力比司维拉姆高200多倍;在pH为5~7的条件下,碳酸镧与磷的结合力比司维拉姆高4倍。
口服碳酸镧的镧生物利用度很低(接近0.001%),吸收的镧主要经胆汁通过肠道排泄,经肾脏排泄的不足2%,因此碳酸镧的血清水平和药代动力学在健康人和CKD5期患者中类似。大多数镧与血浆蛋白结合,血清游离镧浓度<3ng/L。这些特性大大减少了系统暴露、组织分布和发生不良反应的可能性。对使用碳酸镧治疗长达6年的患者进行的研究发现,血浆镧浓度在治疗期间及治疗后都没有增加。镧可在骨和肝脏中极微量沉积,在停止治疗后,骨组织中的镧每年降低约13%。长期使用碳酸镧未发现对肝脏的毒性作用。对认知功能没有影响。镧既不是细胞色素P450酶的底物也不是它的抑制剂,故不会被代谢。碳酸镧也有一定的副作用,与肾源性系统纤维化有关,而且成本也高,可能限制其应用。
近来发现烟酸及烟酰胺可以降低血磷,主要是抑制小肠磷的吸收,其机制就是抑制NaPi-IIb。用量为500mg/d,大大超过日常需要量。势必加剧烟酸及烟酰胺的副作用及依从性。
对于一些高血磷,只能血液透析。每次能降800mg,但血液透析后上升,直到下次血液透析达最高水平。通常可以增加透析频率,每天透析,夜间透析或隔日长时间透析。
尽管各种磷结合剂有各种不良副作用,目前仍然是主要的方法。尤其对于维持性透析者,由于常规透析清除有限和残余肾功能显著下降,大部分需要同时服用磷结合剂,目前新型的磷结合剂以成为主要的药物。
高血磷与慢性肾脏病及血液透析的关系研究相当透彻。患者对高血磷控制的重要性不够理解,饮食依从性差,导致血磷的管理面临极大挑战。因此需要进行各方面的教育及培训,共同解决CKD患者的血磷管理。而低血磷症也不被人们重视。
低磷血症
患者34岁男,因腿及背部虚弱无力越来越重就诊。做一次仰卧起坐也不能,蹲下后就不能起来。血钙正常,血磷0.52mmol/L。24小时尿排出的磷是正常的2倍多,25-(OH)D3正常,而1,25-(OH)2D3检测不到。
问题1患者有虚弱无力、低血磷症,尿磷排出明显增多,考虑哪些疾病?
绝大部分磷在细胞内液,细胞外液磷只占很少部分。当体内缺乏磷时,血磷可能正常。相反,当血磷明显下降时,细胞内可能相对正常。血清磷浓度在0.8-0.96为轻度低磷血症,0.3-0.8为中度低磷血症,0.3以下为重度低磷血症。从下表可知引起低磷血症的原因很多。
低血磷并不罕见,临床上至少有0.43%的患者有严重的低血磷,临床可以出现无力,肌肉瘫痪、昏迷,甚至死亡。重度的低血磷症与横纹肌溶解、溶血、呼吸衰竭及心力衰竭有关。严重的低血磷症增加死亡率。
根据本例资料考虑肾脏排磷增多,即肾小管重吸收磷障碍。甲状旁腺功能亢进及继发性甲状旁腺功能亢进症可以促进尿磷排泄,由于肠道及骨骼的动员可以抵消这一过程,因此甲状旁腺功能亢进,血磷一般不低于0.6mmol/L,除非口服磷结合剂或磷摄入减少。肾脏磷排泄增多需与以下四种疾病鉴别:X连锁低磷酸盐血症、常染色体显性遗传低磷血症佝偻病、肿瘤相关性骨软化症和纤维性结构不良症。
本病由于缺乏1α-羟化酶导致活性维生素D缺乏。活性维生素D低会导致PTH升高。活性维生素D生成增多,促进肠道钙磷吸收可以维持血钙稳定,动员骨钙入血,骨矿化可能不足。但PTH也有抑制肾小管对磷的重吸收,导致尿磷排出增多。
问题2导致低磷血症的机制?
1. 摄入减少,单纯摄入减少很少引起低磷血症,因为肾脏会相应增加磷的重吸收。如存在磷吸收不良或使用磷结合剂有可能导致血磷下降。
2. 细胞内外转运
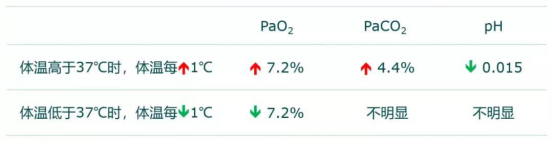
主要见于呼吸性碱中毒及再喂养综合征。呼吸性碱中毒时由于PH升高,导致磷酸果糖激酶活化,刺激糖酵解,消耗磷酸盐,导致低磷。另外由于PaCO2下降有可能抑制PTH的作用,导致排磷减少、重吸收增加,肌肉对磷的摄入也增加。再喂养综合征患者低磷血症的发生与进食后胰岛素过多释放引起磷酸盐细胞内转移有关。ATP,2,3-二磷酸甘油酸盐、肌酸磷酸激酶合成增多也导致了再进食综合征低磷血症的发生。
3. 排磷增加
这是低磷血症的主要原因。影响肾脏排磷的有PTH,FGF32等。PTH增加可以增加排磷,FGF32过多表达,导致NaPiII转运体降解。
低血磷佝偻病由于各种遗传性或获得性病因导致肾脏排磷增多,引起以低血磷为特征的骨骼矿化障碍性疾病,具有高的致残率、致畸率。发生在儿童为佝偻病,方颅、鸡胸、肋骨串珠、四肢弯曲畸形(O形或X形腿),生长迟缓。成人起病称为骨软化症,表现为乏力、体型改变、身材变矮、多发骨折、骨痛等。
遗传性 XLHR(PHEX) ADHR(FGF23) ARHR(DMP-1,ENPP1) HRHPT(低血磷性佝偻病合并甲状旁腺功能亢进(KLOTHO 易位) McCune-Albright综合征(MAS)(GNAS) 颅面骨发育不良 (OGD) 获得性 肿瘤性骨软化症(TIO) 范可尼综合征
|
X-连锁低磷性佝偻病由于PHEX基因突变所致。定位XP22.11,PHEX对FGF23的抑制表达作用减弱,导致FGF23活性增加。从而出现低血磷。还表现为身材矮小、O型腿、FGF23水平升高,1,25(OH)2D3正常或降低。